- Research article
- Open access
- Published:
A molecular epidemiological study on Escherichia coli in young chicks with colibacillosis identified two possible outbreaks across farms
Veterinary Research volume 54, Article number: 10 (2023)
Abstract
Avian pathogenic Escherichia coli (APEC) is the cause of colibacillosis outbreaks in young poultry chicks, resulting in acute to peracute death. The high morbidity and mortality caused by colibacillosis results in poor animal welfare, reduced sustainability and economical loss worldwide. To advance the understanding of the molecular epidemiology, genomic relatedness and virulence traits of APEC, we performed systematic sampling from 45 confirmed colibacillosis broiler flocks with high first week mortality (FWM) during 2018–2021. From these flocks, 219 APEC isolates were whole genome sequenced (WGS) and bioinformatic analyses were performed. The bioinformatic analyses included sequence typing (ST), serotyping, detection of virulence-associated genes (VAGs) and phylogenetic analysis. Our results showed a high prevalence of ST23, ST429 and ST95 among APEC isolates from Norwegian broiler flocks, and identified ST23, ST429, ST117 and ST371 to cause disease more often alone, compared to ST95, ST69 and ST10. Phylogenetic analyses, together with associated metadata, identified two distinct outbreaks of colibacillosis across farms caused by ST429 and ST23 and gave insight into expected SNP distances within and between flocks identified with the same ST. Further, our results highlighted the need for combining two typing methods, such as serotyping and sequence typing, to better discriminate strains of APEC. Ultimately, systematic sampling of APEC from multiple birds in a flock, together with WGS as a diagnostic tool is important to identify the disease-causing APEC within a flock and to detect outbreaks of colibacillosis across farms.
Introduction
Colibacillosis is considered a disease of high importance in poultry production as it gives rise to poor animal welfare and high economic losses worldwide. It is caused by avian pathogenic Escherichia coli (APEC) which is classified as an extra-intestinal pathogenic E. coli (ExPEC), together with neonatal meningitis-causing E. coli (NMEC) and uropathogenic E. coli (UPEC). Colibacillosis is suspected when the mortality among young chicks in a flock increases rapidly during the first week after hatching, usually peaking between two to 5 days after hatching. At necropsy, the chicks show typical macroscopic lesions associated with acute to per acute polyserositis, such as an enlarged spleen, edematous serous membranes and umbilicus. At a later stage of the disease, colisepticemia may lead to fibrinous polyserositis with fibrin exudates covering the serosa of the inner organs like the liver (perihepatitis) and heart (pericarditis). Isolation and identification of E. coli confirms the diagnosis [1].
Extra-intestinal pathogenic E. coli, including APEC, is considered a diverse group of pathogens, and several studies have attempted to group and identify APEC according to virulence-associated properties [2,3,4]. Historically, E. coli has been serogrouped by detecting somatic O–antigens with antisera. The most common APEC serogroups are O1, O2, and O78 [1, 5]. Later on, phylogrouping, a triplex PCR method to group E. coli based on the presence or absence of three genes divides E. coli into seven phylogroups: A, B1, B2, C, D, E and F, have been used [6]. MLST is another common method for typing E. coli, including APEC, and it is based on a combination of seven housekeeping genes in the E. coli genome [7]. Some of the most common APEC STs identified in Europe include ST23, ST69, ST95, ST117, ST131, ST140 and ST428/429 [5].
Whole genome sequencing (WGS), on the other hand, enables an array of in silico characterization methods, such as MLST, serotyping, characterization of VAGs and phylogenetic analysis [5, 8]. Core gene analysis and phylogenetic analysis, detect variations at nucleotide level and may be used to study relatedness, the latter being useful to detect and confirm possible outbreaks and their origin. Phylogenetic methods, however, are continuously developing and the outcomes of such molecular analyses require in-depth genomic understanding, essential bioinformatic knowledge as well as understanding of the pathogens to be studied [9, 10].
Between 2014 and 2016 the Nordic countries, including Norway, experienced a sudden increase in flocks diagnosed with colibacillosis [11]. Whole genome sequencing and genomic investigation of isolates from the affected flocks revealed a predominant lineage of ST117 O78:H4, but also a genetically diverse population. The experience highlighted the need for more knowledge of the molecular epidemiology of APEC in the Norwegian broiler production. In 2018, a systematic sampling of broiler flocks with high first week mortality (FWM) was therefore initiated.
The aim of this study was to systematically collect APEC isolates from Norwegian broiler flocks with high FWM and use WGS and bioinformatic analysis for in depth characterization and comparison of isolates from the same flock and between flocks. Flock-related metadata such as sampling date, hatchery, parent-flocks and hybrid were collected at each sampling occasion, enabling detection of potential association between APEC types identified. Finally, the relationship between the identified STs and their serotype- and VAG- profiles were studied.
Materials and methods
Study design: Necropsy and sampling
Flocks with predicted FWM above 2% elicited sampling from a poultry veterinarian. The flocks were from different regions of Norway and of different hybrids, though the predominant hybrid in Norway is at present Ross 308 (Table 1). Necropsies on ten birds/flock, recently died or euthanized for animal welfare reasons, were performed by a poultry veterinarian. Macroscopic lesions were noted as present/absent in a predefined submission form. Five of the ten examined birds with the most typical macroscopic lesions associated with colibacillosis were selected for bacteriological examination from the spleen, liver and one other organ as previously described in Kravik et al. [12].
Bacteriological examination
Each sample was streaked onto two blood agar (BA) plates and one heart infusion agar (HIA) and incubated at 37 °C anaerobically, in a CO2 chamber and under normal atmospheric pressure, respectively, according to standard procedures for bacterial diagnostics as described in Kravik et al. [12]. After 18–24 h of incubation all samples were examined for the presence of E. coli and the colony morphology on the three agar plates were described. Bacterial growth was divided into sparse, medium, or rich, and the level of purity of E. coli was graded from 1 to 4: Pure growth (1), almost pure growth (2), dominating growth of E. coli (3) and mixed culture (4). A grade 2 was given if a few colonies of Enterococcus spp. or Proteus spp. were present on the agar, together with an otherwise pure culture of E. coli. Grade 3 was defined by dominating growth of E. coli, but in combination with sparse to medium growth of either Enterococci spp., Proteus spp. or less growth of a different bacterium. A mixed culture, grade 4, was defined based on growth of a minimum of three different bacteria, where E. coli was not the dominating bacterium on the three agar plates (Additional file 1). During bacteriological examination, at least one confirmed E. coli isolate from each organ was frozen and stored for future analysis.
Flock diagnosis
An individual diagnosis of colibacillosis was given based on the presence of pathological lesions typically associated with colibacillosis septicemia in combination with a bacteriological examination graded 1–3. We defined a flock diagnosis if FWM was higher than 1.5% and at least three out of the five sampled birds from the flock were diagnosed with colibacillosis individually.
Whole genome sequencing
From each flock with a confirmed colibacillosis diagnosis, 3–5 birds per flock were sampled and one isolate from each sampled bird was selected for WGS. Each isolate was preferably isolated from the liver and DNA was extracted as described previously [12]. Genomic DNA samples from 204 isolates were prepared and sequenced at the Norwegian Veterinary Institute (NVI). An additional 15 isolates were included, previously described in Kravik et al. [12]. All 219 isolates were subjected to library preparation: Nextera™ DNA Flex library preparation (Illumina), and sequenced on an Illumina MiSeq instrument, resulting in 300 bp paired-end reads. The sequence data analyzed in this study are found publically available in the ENA database with bioprojects PRJEB43441 and PRJEB55163. See Additional file 1 for individual accession numbers.
In silico analysis
Whole genome sequence assembly and typing
The Bifrost pipeline [13] was used for initial quality control and assembly. This pipeline consists of read quality control, trimming, removal of PhiX and assembly. ARIBA [14] version 2.14.6 was used to determine the sequence types (ST) according to the Achtman scheme [7]. Isolates with novel sequence types were uploaded in Enterobase for ST assignment [15]. Serotypes were identified using SerotypeFinder [16] version 2.0.2.
Virulence-associated genes
Analysis for detection of VAGs was performed using VirulenceFinder version 2.0.4. The VirulenceFinder database was extended by adding known APEC-associated genes found in the Virulence Factors of Pathogenic Bacteria database (Additional file 2), [2, 17,18,19]. The complete database consisted of 629 entries of virulence-associated genes and their variants.
Core gene analysis and phylogenetic analysis
All isolates that passed QC parameters were included in a phylogenetic analysis based on the core genes. The ALPPACA pipeline [20] version 1.0.0 was used to run genome annotation with Prokka [21] version 1.14.6, followed by pangenome analysis with Panaroo [22] version 1.2.9 to detect and align the core genes among the 219 genomes. Constant sites were removed from the alignment by using Snp-sites [23] version 2.5.1. Snp-dists [24] and version 0.8.2 was used to calculate the pairwise SNP distances from the alignment. Lastly, IQTree [25] version 2.1.4 was used to generate the phylogenetic tree, using Ultrafast bootstrapping [26] with 1000 replicates, and model finder plus [27] for model selection.
Within two of the most frequent STs (ST23 and ST429) separate phylogenetic analyses with ALPPACA were performed. ParSNP [28] version 1.6.1 was used to generate a core genome alignment, followed by detection of recombinant regions with Gubbins version 3.2.0 using RaxML as the treebuilder and the GTRGAMMA model. Maskrc-svg [29] version 0.5 was subsequently used to mask recombinant areas from the alignment. Constant sites were removed by using Snp-sites, followed by pairwise SNP distance calculation with snp-dists and phylogenetic inference with IQTree, similar to above. All phylogenetic trees were visualized in R [30] version 4.0.2, using the ggtree package version 3.0.4 [31].
Results
Sampling and colibacillosis confirmation
From September 2018 to June 2021, 45 broiler and four broiler breeder rearing flocks were sampled, resulting in a total of 49 flocks with FWM ranging from 1.53% to 12.6%. The flocks were of different hybrids, all less than 14 days of age, from various regions in Norway and originated from three different hatcheries. Four broiler flocks were not diagnosed with colibacillosis, and thereby excluded from further analysis. Altogether, 45 flocks with high FWM were given a colibacillosis diagnosis and further included in the analysis (Table 1).
Whole genome sequencing and in silico analysis
Quality control
Altogether 219 confirmed E. coli isolates were sequenced. One E. coli was selected from each bird, and 3–5 birds were sampled from 45 flocks (Additional file 1). Initial quality control of the genome sequences, based on multiQC and Quast report, showed that the GC content of the isolates were between 50.51 and 50.57%, the number of contigs were 43–74 and the total length of the complete genome after assembly was 4.86–4.96 Mbp (See individual quality scores for all isolates in Additional file 1).
MLST and serotyping
The 219 APEC isolates from 45 flocks were sequenced and characterized by ST and in silico serotype. Twenty-six of these flocks exhibited the same ST in all isolates from within a flock, while 35 flocks were identified with up to two STs and therefore identical ST in a minimum of three birds from within the same flock (Table 1 and Additional file 1). Ten of the 45 flocks were identified having three or more STs within the flock (Table 1).
Altogether 32 different STs were identified, of these, 15 were only identified once. The most common STs identified in this study were ST23, ST429, ST95, ST117, ST371, ST69 and ST101 in descending order (Table 2).
In silico serotyping revealed a total of 38 distinct serotypes. Some serotype profiles were detected within a single ST, whereas other serotype profiles were detected across several STs. Of the ten most common STs, five STs were identified with several serotype profiles. The most common serotypes were O1:H7, O2/O50:H1, O2/O50:H5, O45:H19 and O78:H4 (Table 2).
Phylogenetic analysis
To investigate the relationship across STs and between STs and serotypes, a core gene analysis of all 219 isolates was performed (Figure 1). Among the 219 isolates, the pangenome analysis detected 14,332 unique genes. Out of these, 3303 were defined as core genes as they were present in at least 95% of the genomes. Model finder plus identified GTR + F + I + G4 as the best-fitting model. The phylogenetic tree revealed that the isolates clustered according to their STs and it showed that isolates with the same serotype profile might be genetically distant. Isolates of serotype O78:H4 were identified as both ST23 and ST117, and serotype O2/O50:H1 was identified as both ST429 and ST135. Further, ST95 and ST117 also contained several serotype profiles: O1:H7 and O2/O50:H5 and O24:H4, O78:H4 and O161:H4, respectively.

Maximum likelihood core-gene SNP tree of all isolates included in the study. Bootstrap values above or equal to 95 are denoted as black nodes. The core gene tree visualizes the genetic relations between the most common sequence types (STs) identified and what serotype profiles are linked to these. Colors on the tips and clade labels represent serotypes and STs, respectively, represented by more than five isolates. Tip labels represent flock and bird.
The two largest clusters in the core gene tree were represented by ST23 and ST429, respectively. Both STs had one distinct serotype profile and all flocks, except one flock with ST429, clustered according to hybrid, sampling dates and ST (Figure 1, Table 1). These two STs were therefore suspected to represent two outbreaks of colibacilllosis and separate phylogenetic analyses were therefore carried out for each of them (Figures. 2 and 3).

Maximum likelihood core genome tree visualizing the genetic relations of all isolates identified as ST23 from 17 flocks (n = 81 isolates). Bootstrap values above or equal to 95 are denoted as black nodes. Tip labels represent flock and bird.

Maximum likelihood core genome tree of all isolates identified as ST429 from seven flocks (n = 33 isolates). Bootstrap values above or equal to 95 are denoted as black nodes. Tip labels represent flock and bird. Colors on the tips represent hybrid. Clade A and C consists of isolates from three flocks each, all of hybrid Ross 308. Clade B represents a single Sasso flock.
A total of 81 isolates from 17 flocks were confirmed to be of ST23. All ST23 isolates were collected from broiler flocks of the hybrid Ross 308 and sampled from March 2021 until the end of May 2021 (Table 1). The phylogenetic analysis of ST23 had an average genome coverage of 94.0%, a SNP range of 0–33 and a mean and median SNP distance of 15. Individual isolates of ST23 clustered, for the most part, according to individual flocks, but there were also isolates which clustered with isolates from different flocks (Figure 2 and Table 3). Within a flock, all but two flocks had mean SNP distances below 10. The SNP range varied from 0 to 1 (smallest range) to 0–33 (highest range) within a flock (Table 3).
ST429 was identified in 33 isolates from seven flocks; six flocks of the hybrid Ross 308 sampled between September 2018 to January 2019, and one flock of the hybrid Sasso sampled in August 2019. The phylogenetic analysis revealed an average genome coverage of 95.8% and the SNP range was 0–172 with a median SNP distance of 62. The tree diagram shows that ST429 isolates separate into three dominating clades: ST429-A, ST429-B and ST429-C (Figure 3). Isolates from the only Sasso flock clustered together in clade ST429-B. Isolates in clade ST429-A and ST429-C originated from Ross 308 flocks from different geographical locations, hatcheries and parent flocks, however, all Ross 308 broiler rearing flocks are distributed from one main hatchery (Figure 3). Within the individual flocks identified with ST429, the mean SNP distances were in all cases < 10 SNPs, except from the Sasso flock, and the SNP range varied from 0 to 1 to 4–21 (Table 3).
The phylogenetic analysis of ST429 was re-run, including only flocks of hybrid Ross 308. The results from this analysis showed an average genome coverage of 95.7% and the SNP range was 0–76 with a median SNP distance of 59 (Additional file 3).
Virulence-associated genes
From a database containing 629 entries of VAGs, 112 VAG-variants were identified in at least one of the APEC isolates from the present study (Additional file 1). The frequency of VAGs was described separately for isolates of ST23 and ST429 and a third group that included all other isolates (Figure 4).

Frequency of virulence-associated genes (VAGs) present in three groups of isolates: ST23 (n = 81), ST429 (n = 33) and a third group comprising the remaining isolates (n = 105). The colors represent the frequency from which each gene is present: red = 100%, black = 0% and yellow = 1–99%. The VAGs are categorized and described as previously presented by Nolan et al. [1] and Kathayat et al. [18].
A total of 25 VAGs were identified in all isolates of ST23, while five VAGs were identified in only some of the ST23 isolates. These include traT, cba, cia and cma that were identified in only 28%, 17%, 12% and 17% of these isolates, respectively. The gad gene was identified in 94% of all the ST23 isolates (Figure 4). Thirty three of the VAGs were present in all isolates of ST429, while four genes were found in some isolates. The tsh, sitC, vat and gad genes were identified in 61%, 94%, 12% and 30% of the ST429 isolates, respectively (Figure 4).
In the third group containing all other STs, there was higher diversity in the presence of VAGs. Similar to ST23 and ST429, all of the isolates in the third group carried the genes csgA-F, ecpA, ibeB, ibeC, iucC, sitD and terC. Several other VAGs were present in more than 90% of all the isolates in the third group, but there were also VAGs that were absent or only present in a few isolates (Additional file 1 and Figure 4). The siderophore receptor-genes fyuA and irp2 were present in all ST23 and ST429 isolates, but only present in 51% and 52% of the other isolates, respectively.
Discussion
There is limited knowledge about APEC in the Norwegian broiler production, and after the peak of outbreaks of colibacillosis on farms in the Nordic countries in 2014, we saw the need for a systematic study of the epidemiology of APEC in the Norwegian broiler production. To identify APEC types within the Norwegian broiler production, we performed systematic sampling of flocks with high FWM and sequenced the genomes of individual isolates from these flocks to study their STs, serotype profile, content of VAGs and their evolutionary relationship.
The selection of isolates was carried out according to the results from a pilot project in 2019. The study concluded (1) low diversity of APEC within individual diseased birds, and (2) the need of sampling a minimum of three birds per flock to identify the diversity of APEC within a flock and determine the main disease-causing APEC at flock level [12].
Out of the 32 distinct STs identified in the present study, almost 50% were only identified once. A similar trend has been observed in previous studies [32,33,34]. The high number of STs only represented by one or two isolates influences the high diversity of STs reported. Whether these single ST strains are true avian pathogens and important for the colibacillosis etiology, or sporadic findings without importance for pathogenicity, is currently not known. These reports, however, highlight the need for sampling multiple animals and WGS of several isolates from a flock to identify the main disease-causing APEC within a flock.
Altogether, 35 of 45 diseased flocks were identified with a dominating APEC type, and ST23, ST117, ST371 and ST429 were more often found to cause disease in a flock alone than other STs. The latter indicates that these STs are possibly more pathogenic in poultry, compared to the STs more commonly identified in mixed infections with more than one ST within a flock [35]. Altogether, ten flocks exhibited a combination of several STs and the APEC types most commonly identified in combination with other STs were ST10, ST69, ST95 and ST101.
Notably, both ST69 and ST95 are frequently isolated in human infections [5, 36]. However, due to their presence in a majority of mixed infections, it is likely to assume that they are rather opportunistic than highly pathogenic in poultry. This is supported by Kromann et al. where ST95 was identified with the highest prevalence from healthy poultry flocks sampled in a non-outbreak situation [37].
Caution should be taken, however, in the discussion of ST95 as this ST is identified with several serotype profiles, and variants of the same ST but with different virulence properties, exists [5, 36, 37]. Our study shows further examples of STs with several serotype profiles, such as ST101 where O88:H8 was identified from three flocks and O103:H21 from another flock and ST117, which exhibits three serotypes O24:H4, O78:H4 and O161:H4 from three different flocks. Serotype O78:H4 is the same serotype as found in ST23. Generally, typing of APEC should be evaluated with caution, as shown in the core gene analysis where ST117 and ST23 exhibit large genetic distances between these STs, even though they share one of the same serotype profiles. Without WGS data, the two distinct peaks of ST117 in 2014 [11] and ST23 in 2021, with possible distinct origins, would be considered the same APEC if only serotyping was performed, but two distinct APEC types if MLST was performed. Sequence types identified with several serotype profiles, on the other hand, suggests that a combination of two typing methods provides better differentiation between APEC isolates (for quick diagnosis in outbreak situations) [35].
Even though APEC is considered a diverse pathogen, there is less variation amongst the most frequent STs reported [12, 33, 35, 38,39,40]. The high prevalence of certain STs, however, may be due to small outbreaks of colibacillosis from a single source and within a limited period, possibly from higher up in the production pyramid [41].
Phylogenetic analysis is today considered the gold standard to evaluate relatedness between isolates from a possible single source. Accordingly, this study presents the mean SNP-distance within, and between flocks identified with the same ST. The results are valuable for the evaluation and understanding of future outbreaks of colibacillosis across farms. To our knowledge, there is no consensus for defining the acceptable number of SNPs in APEC outbreaks [9, 10, 12]. Factors such as the mutation rate, the number of individuals the pathogen might encounter, pathogen pressure and the duration of source contamination could influence the number of SNPs emerging during an outbreak [9]. The consideration of SNP distances between isolates from an outbreak, should therefore, include the pyramidal structure of the poultry production and the number of individuals and generations the pathogen will encounter in a potential vertical transmission line, before causing disease in a broiler chick. Further, SNP distances alone should not be assessed without the knowledge of the proportion of the genome that has been used in the analysis. Therefore, pathogen- and population metadata, as well as genome coverage, plays as much a role in the understanding if an isolate belongs to an outbreak as the SNP distance alone [9, 10, 12].
Altogether, 112 VAGs were identified at least once in our set of APEC isolates. This correlates well with recent reports by Apostolakos et al., who identified 113 VAGs in their study [32]. However, within each of the two main STs (ST23 and ST429) identified in the present study, there was, with few exceptions, less diversity. This is expected as the isolates within these STs were considered part of the same outbreaks, and therefore likely to be clonal. The genes varying within ST23 were traT, cba, cia and cma, and within ST429 vat and gad. Most of these genes are known to be plasmid-encoded and might therefore vary more frequently between isolates within an outbreak than chromosomally encoded genes. Further, fyuA and irp2 were represented in all outbreak isolates, but only in approximately half of the non-outbreak isolates. Both these VAGs belong to the Yersiniabactin operon which is responsible for iron acquisition and of high importance in the pathogenesis of avian colibacillosis [1].
A few VAGs were, on the other hand, identified in all isolates in this study and could therefore be considered important for the virulence of APEC. However, without a true, non-pathogenic control group for comparison, no valid conclusions may be drawn based on these data as these VAGs might be identified in all avian E. coli isolates regardless of pathogenicity. A recent study from Johnson et al. [35] suggested two conserved VAGs associated with APEC plasmid, hlyF and ompT as potential markers for increased virulence potential in combination with other genetic features. In the present study, hlyF and ompT were identified in all ST23 and ST429 isolates and in 89 and 96% of isolates of other STs, respectively.
In this study, the virulence of an APEC strain was evaluated based on the prevalence of the identified ST and its’ ability to cause high FWM within a flock and across farms alone or in combination. Further, we have given insight into the relatedness of outbreak strains, and presented the most prevalent VAGs associated with the two outbreak strains ST23 and ST429 in our study. However, APEC strains emerge from multiple E. coli lineages, and for the future it would be interesting to follow single ST outbreaks to identify if the same ST reappear as more prone to cause outbreaks across farms or if other STs, which in this study appear of lesser importance, could be the cause of future outbreaks. It would be interesting to better identify the transmission routes of the pathogen in outbreak situations between farms. A common database for the control and prevention of APEC outbreaks has been suggested [42]. For the future, the authors supports an initiative for such a database, including well-defined metadata as well as comparable sampling and diagnostic methods. Such a database could aid in the identification of pathogen transmission routes through the broiler poultry pyramid. Further, the comparison of VAGs from systematically sampled outbreak strains to a proper control group would be of interest to further unravel the virulence potential of individual APEC. The importance of defining what an APEC control is, should however, be further discussed as commensal E. coli might have the potential to become APEC and the pathophysiology behind is still not well defined [38].
In conclusion, this study shows the presence and distribution of APEC types identified from local outbreaks of colibacillosis-septicemia across Norway during 2018–2021. Further, it identifies how peaks of high FWM due to colibacillosis may be caused by a single, distinct ST. Phylogenetic analysis gives insight into the relatedness between isolates belonging to the same ST, but also across STs and serotypes, identifying the need for combining typing methods to better discriminate between APEC types. This study also highlights the value of using WGS as a diagnostic tool for surveillance as well as in the identification of future outbreaks of colibacillosis.
References
Nolan LK, Vaillancourt J-P, Barbieri NL, Logue CM (2020) Colibacillosis. In: Swayne DE, Boulianne M, Logue CM, McDougald LR, Nair V, Suarez DL, Wit S, Grimes T, Johnson D, Kromm M, Prajitno TY, Rubinoff I, Zavala G (eds) Diseases of Poultry. Wiley Blackwell, New York
Ewers C, Janßen T, Kießling S, Philipp H-C, Wieler LH (2005) Rapid detection of virulence-associated genes in avian pathogenic Escherichia coli by multiplex polymerase chain reaction. Avian Dis 49:269–273
Rodriguez-Siek KE, Giddings CW, Doetkott C, Johnson TJ, Nolan LK (2005) Characterizing the APEC pathotype. Vet Res 36:241–256
Johnson TJ, Wannemuehler Y, Doetkott C, Johnson SJ, Rosenberger SC, Nolan LK (2008) Identification of minimal predictors of avian pathogenic Escherichia coli virulence for use as a rapid diagnostic tool. J Clin Microbiol 46:3987–3996
Mehat JW, van Vliet AHM, La Ragione RM (2021) The Avian Pathogenic Escherichia coli (APEC) pathotype is comprised of multiple distinct, independent genotypes. Avian Pathol 50:402–416
Clermont O, Christenson JK, Denamur E, Gordon DM (2013) The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep 5:58–65
Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M (2006) Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151
Lozica L, Repar J, Gottstein Ž (2021) Longitudinal study on the effect of autogenous vaccine application on the sequence type and virulence profiles of Escherichia coli in broiler breeder flocks. Vet Microbiol 259:109159–109159
Duval A, Opatowski L, Brisse S (2022) Is it the same? straiN Defining genomic epidemiology thresholds tailored to individual outbreaks. bioRxiv. https://doi.org/10.1101/2022.02.15.480545
Schürch AC, Arredondo-Alonso S, Willems RJL, Goering RV (2018) Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene-based approaches. Clin Microbiol Infect 24:350–354
Ronco T, Stegger M, Olsen RH, Sekse C, Nordstoga AB, Pohjanvirta T, Lilje B, Lyhs U, Andersen PS, Pedersen K (2017) Spread of avian pathogenic Escherichia coli ST117 O78:H4 in Nordic broiler production. BMC Genomics 18:13
Kravik IH, Kaspersen H, Sjurseth SK, Jonsson M, David B, Aspholm M, Sekse C (2022) High sequence similarity between avian pathogenic E. coli isolates from individual birds and within broiler chicken flocks during colibacillosis outbreaks. Vet Microbiol 267:109378
Norwegian Veterinary Institute/Bifrost: Specific genes finding in working order, https://github.com/NorwegianVeterinaryInstitute/Bifrost
Hunt M, Mather AE, Sanchez-Buso L, Page AJ, Parkhill J, Keane JA, Harris SR (2017) ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genom 3:e000131
Zhou Z, Alikhan N-F, Mohamed K, Fan Y, Achtman M (2020) The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res 30:138–152
Joensen KG, Tetzschner AM, Iguchi A, Aarestrup FM, Scheutz F (2015) Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J Clin Microbiol 53:2410–2426
Virulence factors of pathogenic bacteria, Escherichia, http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi
Kathayat D, Lokesh D, Ranjit S, Rajashekara G (2021) Avian pathogenic Escherichia coli (APEC): an overview of virulence and pathogenesis factors, zoonotic potential, and control strategies. Pathogens 10:467
Newman DM, Barbieri NL, de Oliveira AL, Willis D, Nolan LK, Logue CM (2021) Characterizing avian pathogenic Escherichia coli (APEC) from colibacillosis cases, 2018. PeerJ 9:e11025
Norwegian Veterinary Institute, ALPPACA, First full release, (https://zenodo.org/record/6323152#.YwTGIdgzbcshttps://zenodo.org/record/6323152#.YwTGIdgzbcs)
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SDW, Corander J, Bentley SD, Parkhill J (2020) Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol 21:180
Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, Harris SR (2016) SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom 2:e000056
Github, tseemann, snp-dists, https://github.com/tseemann/snp-dists
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2017) UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol 35:518–522
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Treangen TJ, Ondov BD, Koren S, Phillippy AM (2014) The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524
Github, kwongj, maskrc-svg, https://github.com/kwongj/maskrc-svg
The R project for statistical computing, https://www.R-project.org/
Yu G (2020) Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics 69:e96
Apostolakos I, Laconi A, Mughini-Gras L, Yapicier ÖŞ, Piccirillo A (2021) Occurrence of colibacillosis in broilers and its relationship with avian pathogenic Escherichia coli (APEC) population structure and molecular characteristics. Front Vet Sci 8:737720
Cummins ML, Reid CJ, Roy Chowdhury P, Bushell RN, Esbert N, Tivendale KA, Noormohammadi AH, Islam S, Marenda MS, Browning GF, Markham PF, Djordjevic SP (2019) Whole genome sequence analysis of Australian avian pathogenic Escherichia coli that carry the class 1 integrase gene. Microb Genom 5:e000250
Delannoy S, Schouler C, Souillard R, Yousfi L, Le Devendec L, Lucas C, Bougeard S, Keita A, Fach P, Galliot P, Balaine L, Puterflam J, Kempf I (2020) Diversity of Escherichia coli strains isolated from day-old broiler chicks, their environment and colibacillosis lesions in 80 flocks in France. Vet Microbiol 252:108923
Johnson TJ, Miller EA, Flores-Figueroa C, Munoz-Aguayo J, Cardona C, Fransen K, Lighty M, Gonder E, Nezworski J, Haag A, Behl M, Kromm M, Wileman B, Studniski M, Singer RS (2022) Refining the definition of the avian pathogenic Escherichia coli (APEC) pathotype through inclusion of high-risk clonal groups. Poultry Sci 101:102009
Jørgensen SL, Stegger M, Kudirkiene E, Lilje B, Poulsen LL, Ronco T, Pires Dos Santos T, Kiil K, Bisgaard M, Pedersen K, Nolan LK, Price LB, Olsen RH, Andersen PS, Christensen H (2019) Diversity and population overlap between avian and human Escherichia coli belonging to sequence Type 95. mSphere 4:e00333-e418
Kromann S, Baig S, Stegger M, Olsen RH, Bojesen AM, Jensen HE, Thøfner I (2022) Longitudinal study on background lesions in broiler breeder flocks and their progeny, and genomic characterisation of Escherichia coli. Vet Res 53:52
Mageiros L, Méric G, Bayliss SC, Pensar J, Pascoe B, Mourkas E, Calland JK, Yahara K, Murray S, Wilkinson TS, Williams LK, Hitchings MD, Porter J, Kemmett K, Feil EJ, Jolley KA, Williams NJ, Corander J, Sheppard SK (2021) Genome evolution and the emergence of pathogenicity in avian Escherichia coli. Nat Commun 12:765
Oikarainen PE, Pohjola LK, Pietola ES, Heikinheimo A (2019) Direct vertical transmission of ESBL/pAmpC-producing Escherichia coli limited in poultry production pyramid. Vet Microbiol 231:100–106
Papouskova A, Masarikova M, Valcek A, Senk D, Cejkova D, Jahodarova E, Cizek A (2020) Genomic analysis of Escherichia coli strains isolated from diseased chicken in the Czech Republic. BMC Vet Res 16:189
Poulsen LL, Thøfner I, Bisgaard M, Christensen JP, Olsen RH, Christensen H (2017) Longitudinal study of transmission of Escherichia coli from broiler breeders to broilers. Vet Microbiol 207:13–18
Christensen H, Bachmeier J, Bisgaard M (2021) New strategies to prevent and control avian pathogenic Escherichia coli (APEC). Avian Pathol 50:370–381
Acknowledgements
This study was funded by the Norwegian Research Council, Research Fund for Agriculture and the Food Industry (NFR project 280385). The bioinformatics analyses were performed on resources provided by UNINETT Sigma2- The National Infrastructure for High Performance Computing and Data Storage in Norway. The authors wish to thank the contributing veterinarians Astrid Søyland Grødem, Lena Hollund Surdal, Mona Nordmark (Den Stolte Hane), Aslak Fitjar Oltedal, Torill Kjensmo, Marielle Wigaard, Skjalg Arne Hansen, Pia Køteles, Elisabeth Kverneland (Nortura), Tove Elin Sande, Sigrid Gosse, Morten Høglien, Miriam Garner (Norsk Kylling) and Kate Norum (Ytterøy kylling). A special thanks also goes to Bjarne Bergsjø, Bereket S. Tesfamichael, Basma Asal and Fiona Valerie Franklin-Alming at the bacteriology laboratory and Cathrine Arnason Bøe and SEQ-TECH for the whole genome sequencing at the Norwegian Veterinary Institute for continuous help and support in the laboratory.
Author information
Authors and Affiliations
Contributions
SKS, BD, MA, CS conceptualized and designed the study. IHK, SKS, CS organized the sampling and the laboratory analysis. IHK, HK analyzed the data. HK, KDR assisted in statistical and bioinformatical methods. IHK, HK, SKS, BD, CS interpreted the data. IHK prepared the first draft of the manuscript. HK, SKS, KDR, BD, MA, CS read, contributed to and approved the final manuscript. All authors read and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Pauline Ezanno
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Metadata linked to each isolate sequenced in this study including flock and sampling data, bacteriological grading, sequencing quality measures, virulence-associated genes identified and accession numbers to ENA.
1The sample ID explains from what flock (first number) and bird (last number) the isolate was sampled. 2The bacterial culture was graded according to purity of growth on three agar plates incubated over night in either normal oxygen pressure, CO2 chamber or anaerobically: grade 1 = pure growth, grade 2 = few colonies of Enterococcus spp. or Proteus spp. were present on the agar, together with an otherwise pure culture of E. coli. Grade 3 = dominating growth of E. coli with sparse to medium growth of either Enterococci spp, Proteus spp. or less growth of a different bacteria, grade 4 = mixed culture: minimum of three different bacteria, where E. coli was not the dominating bacteria. 3The presence of pathological lesions associated with colibacillosis (colisepticaemia) within an individual bird during necropsy, 1 if ≥2 lesions present, 0 if <2 lesions present. 4Each individual bird received a colibacillosis diagnosis (1) if the bird had lesions of colisepticaemia and the bacteriological examination from the individual sample was graded 1-3.
Additional file 2. List of genes included in the extended database of genes uploaded to VirulenceFinder for the identification of virulence-associated genes in this study.
Included in the table is the name of gene, short description of gene and from what database the gene was identified (VirulenceFinder or VFDB).
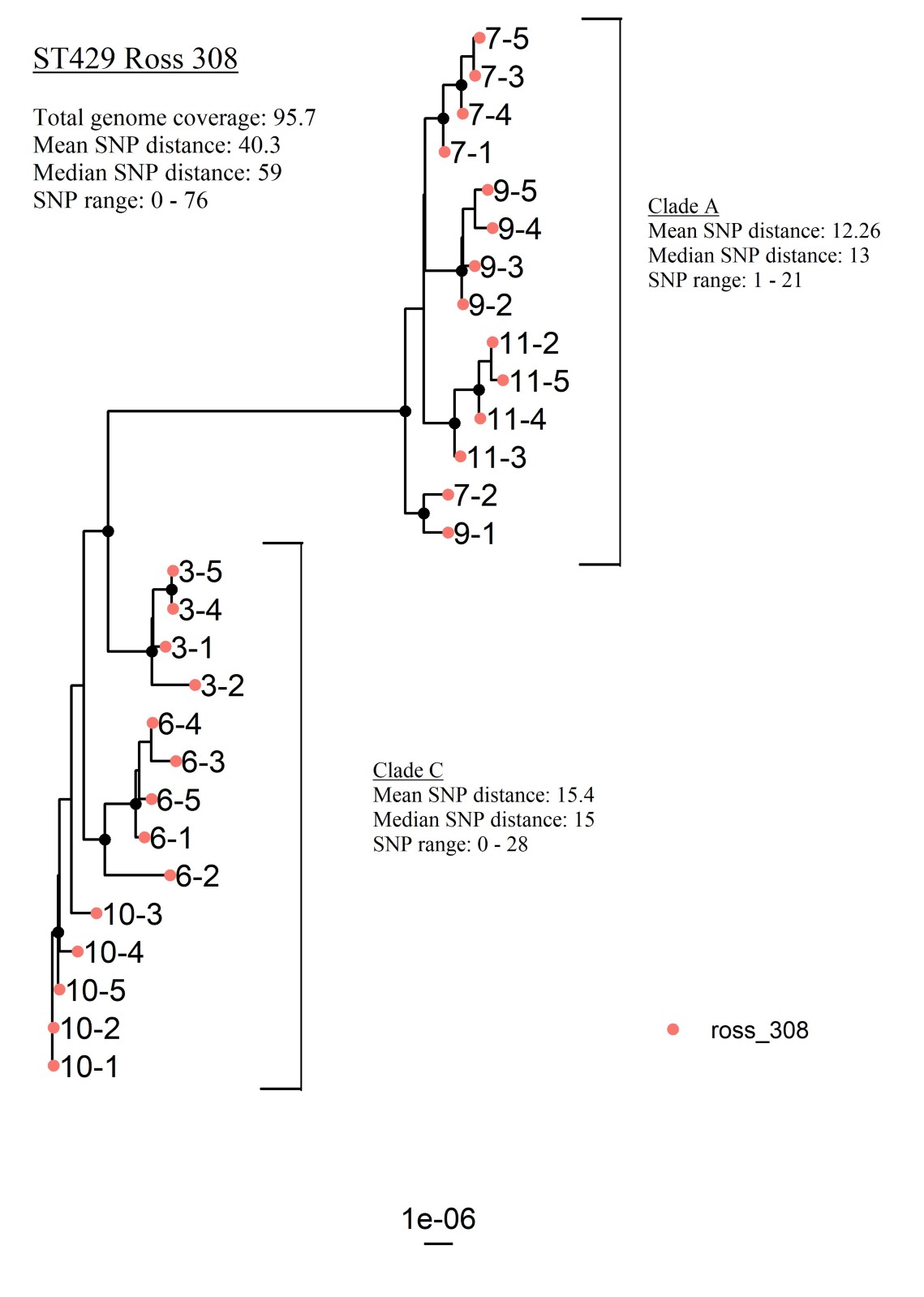
Additional file 3. Maximum likelihood core genome tree visualizing the genetic relations of all isolates identified as ST429 from all Ross 308 flocks (n = 28), excluding isolates from one Sasso flock.
Bootstrap values above or equal to 95 are denoted as black nodes. Tip labels represent flock and bird. Clade A and C consists of isolates from three flocks each, all of hybrid Ross 308.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Kravik, I.H., Kaspersen, H., Sjurseth, S.K. et al. A molecular epidemiological study on Escherichia coli in young chicks with colibacillosis identified two possible outbreaks across farms. Vet Res 54, 10 (2023). https://doi.org/10.1186/s13567-023-01140-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-023-01140-6